Consulting and Training
in pharmaceutical analysis and
Quality Control
Welcome to my website!
I would be pleased to support you, to provide consulting or training with respect to topics of pharmaceutical analysis and Quality Control. My name is Dr. Joachim Ermer and I rely on a long-year experience in pharmaceutical industry.
Following study of biochemistry and PhD thesis in enzyme kinetics at the Martin-Luther-University Halle-Wittenberg in 1988, and a post-doc scholarship in Cambridge, UK, I was employed in various positions in industrial Quality Control since 1991. My special interest has always been analytical validation and method performance as well as related topics such as out-of specification results (OOS), transfer, practical statistics, Quality-by-Design, or lifecycle management. My long-standing participation and discussions in national and international working groups provided me with possibilities to participate as well as to take influence in these developments.
My responsibilities in pharmaceutical analysis included the position of head of laboratory within the analytical drug development at Hoechst AG, Frankfurt, Germany, and from 2001 to 2005 a global function as Director of Analytical Processes and Technology. This comprised consultation, harmonisation, trouble-shooting and training of all industrial sites of Aventis with respect to Quality Control topics, including creation and development of company guidelines.
From 2005 to 2010, I served as head of Quality Control Frankfurt Chemistry, Sanofi, Germany. Between 2010 and 2018 my responsibilities as head of QC Services included a reference standard group with the mission to provide Sanofi-wide management and distribution of analytical reference standards. From 2018 to 2020, as head of QC Lifecycle Management Frankfurt Chemistry, I have evaluated compendial and regulatory changes, supported and coordinated analytical transfers, validation and implementation projects. In order to ensure the suitability and compliance of analytical procedures during the lifecycle, I have developed a quality system for continuous performance verification of all API-methods (monitoring) and accompanied its implementation.

Dr. Joachim Ermer
I hope to have raised your interest and I am looking forward to get in touch with you. Please do not hesitate to contact me for the discussion of topics of your interest:
+49 151 / 28 76 11 66
News
ICH Q2(R2)/Q14 Training Material
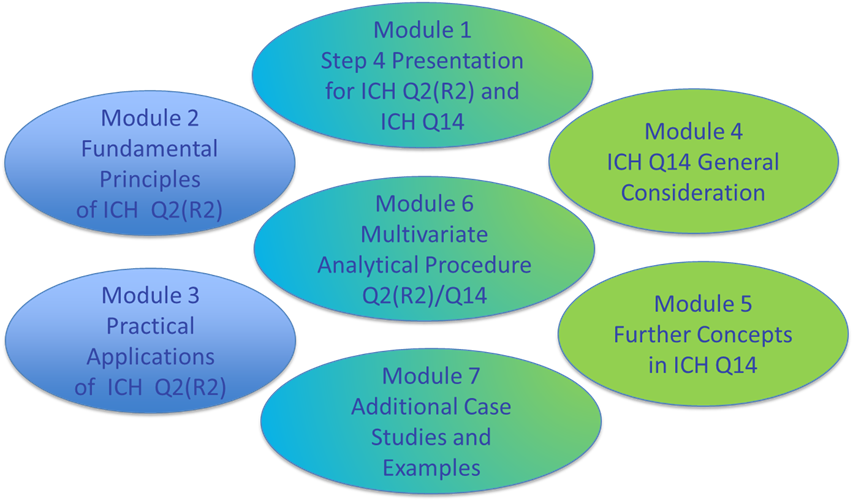
After some delay, in July 2025 the ICH training material was published, with 7 modules.
The training material provides with 400 slides/pages for various aspects additional interpretation and examples, such as
- Analytical Target Profile (Module 2 & 4, examples)
- Platform Analytical Procedures (Module 3: slides 49-55, Module 7: page. 66ff)
- Use of data from development (Module 2, slide 7; Module 3, slides 91ff; Module 7, page 85)
- Calculation of confidence intervals for precision and/or accuracy (simple pooling, Module 3 slides 12, 24, 46, 80)
- Risk assessment (examples Module 7)
Unfortunately, also in the training materials the use of confidence intervals only as acceptance criteria for precision and/or accuracy is discussed (equivalence tests), leaving open conditions justifying the traditional approach comparing the calculated precision and/or accuracy results with the acceptance criteria (point estimates). For my understanding, this is justified in case of simple analytical procedures and less critical Quality Attributes, because here a numerical control of the risk (α-error) is not essentially required. The discussion of an inconclusive evaluation (Module 3, slide 64), i.e. the point estimate within acceptance, the confidence limit(s) outside, is rather weak. (“Evaluate the risk and decide if acceptable per company PQS requirements.”) Interestingly, you will find in the training materials also examples with no calculation of confidence intervals (Module 7, pages 9, 44, 58; Module 3, slide 86).
The almost complete miss to explain how acceptance criteria can be derived is rather disappointing („Provision of acceptance criteria has been deliberately limited“), despite the crucial role for assessing analytical performance. Equally frustrating is the discussion with respect to precision. Here, the various levels are confused and mixed, which does not allow to estimate the variance contributions essential for a since-based establishment of the replication strategy. Although the latter is discussed, any orientation how to derive is missing. Very rightly, the importance of the reportable result (including the replication strategy) is emphasised, but in the examples it is mainly ignored, only intermediate precision is mentioned. However, the latter corresponds just to a single reportable result, excluding any replication to optimise precision by averaging.
The „full factorial validation“ approach described in Module 7 (page 87, table 2) to establish a robustness region (MODR) can be regarded as unrealistic. In case of a full validation of all limits and the setpoint of method parameters, for e.g. 5 parameters with 2 level, already 25+1 = 33 validations would be required. However, the minimum validation of the set-points is also described as an option. But this approach requires at least an assessment in case of changes (which is still much less effort).
The many discussions and examples across the 400 slides/pages will often provide a pragmatic and sensible selection, with reference to the training materials as justification. “In practice, scientific rigor must be applied on a case-by-case basis when determining an appropriate approach or criterion“.
For further information, see: Method Validation in Pharmaceutical Analysis: A Guide to Best Practice, 3rd Edition, Editor(s): Joachim Ermer, Phil Nethercote, Wiley 2025 Print ISBN:9783527348909 28 Online ISBN:9783527831708 |DOI:10.1002/9783527831708 (28 February 2025)
For further information, see:
Validation in Pharmaceutical Analysis – Live Online Training ECA, Course No. 22744
Lifecycle Concept, ICH Q2(R2) & Training Materials, USP <1225> Revision
Wednesday, 13 May 2026 9 .00 – 17.00 h
Draft of a new USP General Information Chapter <1221>: „Ongoing Procedure Performance Verification“
An analytical procedure has to remain fit for its intended purpose throughout its lifecycle, at each application. Of course, the foundation is laid in method development and validation. However, the suitability should be verified across the whole lifecycle. Regulatory guidelines, such as the FDA guidance „Analytical Procedures and Methods Validation for Drugs and Biologics“ (2015), or the ICH Q14 guideline “Analytical Procedure Development” (2023) request a trend analysis and periodic evaluation on method performance to evaluate the need to optimize or to revalidate, but – fortunately – do not go into specific details. This leaves flexibility to try and establish pragmatic approaches. Now, we see further orientation with respect to a systematic monitoring programme with the draft of a new USP General Information Chapter <1221> “Ongoing Procedure Performance Verification” (OPPV) [Pharm. Forum 51.4].
OPPV differs from routine procedure control such as system suitability tests (SST) through its proactive monitoring approach. Data are systematically collected and analysed to identify potential performance issues or adverse trends (e.g. using control charts), to ensure timely corrective actions to prevent significant deviations. An integrated approach to manage and improve analytical procedures with a balanced effort will help to ensure product safety and efficacy alongside business efficiency by fact-based priorisation of resources, by performance-based assessment of suspect (out-of-expectation (OOE), out-of-trend (OOT)), or out-of-specification (OOS) results, and by better discrimination of manufacturing and analytical contributions for a continuous process validation and product assessment.
Stability testing is a fundamental part of pharmaceutical development and quality assurance. It provides evidence on how the quality of a drug substance or drug product varies during the intended shelf-life under the influence of environmental factors such as temperature, humidity, and light. Well-designed stability programs are important to support regulatory submissions, product lifecycle management, and risk-based decision-making in the pharmaceutical industry, with a huge economic impact.
The importance of this topic is illustrated by choosing it as the first one for the international harmonisation process with the ICH guideline Q1A „Stability Testing of New Drug Substances and Products“ (October 1993). In order to take the progress since then into account, in 2022 it was decided to update and revise the stability guidelines. Despite this complex task – which became certainly not easier by the extension of the ICH scope of application – the ICH expert working group has achieved their initial milestones, so that the draft guideline was published for commentation in April 2025.
The new ICH Q1 Guideline draft represents a comprehensive revision and consolidation of the former ICH Q1A-F and Q5C guidelines. The scope was extended to cover both synthetic and biological drug substances and drug products, including vaccines, gene therapies, and combination products. Stability considerations for gene therapy products (e.g. Advanced Therapy Medicinal Products (ATMPs) are newly added in an Annex. The concepts can also be applied to clinical stability investigations, proportionate to the increasing level of understanding during pharmaceutical development, and to reference standards. The new Q1 Draft includes all climatic zones and thus can achieve a real worldwide harmonization. An important extension is the coverage of post-approval changes and the stability life-cycle management, in alignment with the ICH Q12 guideline.
Further new or adjusted topics include:
- Stability requirements for intermediates
- Short-term and in-use stabilities
- Ongoing and lifecycle stability studies
- Statistical analysis of stability data and extrapolation
- Enhanced stability modelling
The principles of Quality by Design and risk-based approaches described within ICH Q8-Q11, and their impact on the overall stability strategy are an important complementation, as for all recent ICH guideline revisions. This is in particular important for alternative and scientifically justified approaches that encompass the variety of different situations that may be encountered.
Considering the complex task and the numerous ICH stake holders, a good balance has been achieved between continuity, adjustments, and modernisation.
For further details, see J. Ermer: Stability Testing of Drug Substances and Drug Products: The New ICH Q1 Draft Guideline. GMP Compliance Adviser, Maas & Peither AG GMP-Verlag (August 2025)